Badania wykazały, że jeden z usuniętych genów powiązanych z zespołem Williamsa jest odpowiedzialny za funkcjonowanie mitochondriów w mózgu
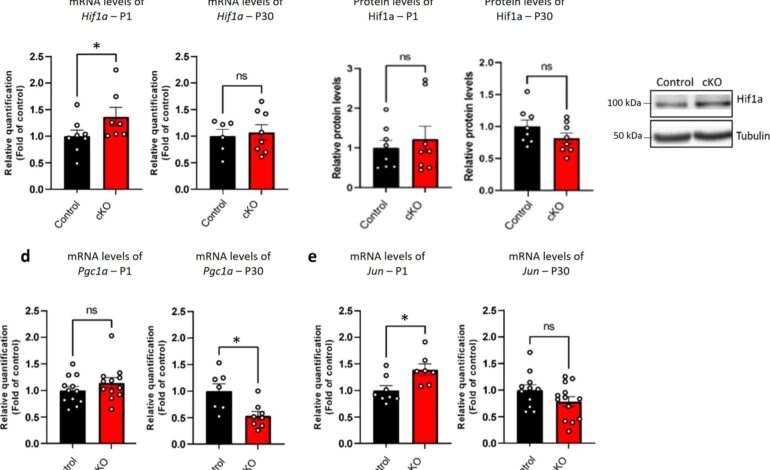
ROS i markery związane z apoptozą są zmienione w całej korze myszy cKO w porównaniu z grupą kontrolną. Zbadano poziomy mRNA genów związanych z ROS i poziomy białek w homogenatach kory myszy cKO w porównaniu z kontrolą. A Właściwości związane z niedotlenieniem zbadano poprzez pomiar Hif1a Poziomy transkryptu mRNA w homogenatach kory od myszy cKO i myszy kontrolnych. W P1 myszy cKO wykazały znacznie wyższe Hif1a Poziomy mRNA (*P = 0,04), ale nie w P30 (ns), w porównaniu z grupą kontrolną. B Homogenaty całej kory z P1 i P30 cKO w porównaniu z próbkami kontrolnymi poddano immunoblotowi z użyciem przeciwciała Hif1a i nie wykazano istotnej różnicy w poziomach białka. C Reprezentatywne wyniki immunoblotu Hif1a. D Właściwości hamowania ROS zbadano poprzez pomiar Pgc1a Poziomy mRNA w homogenatach kory myszy cKO i myszy kontrolnych. W P1 myszy cKO nie wykazały istotnej różnicy (ns), podczas gdy w P30 myszy cKO wykazywały znacząco niższy poziom ekspresji Pgc1a w porównaniu do kontroli (*P = 0,01). mi Właściwości apoptotyczne badano poprzez pomiar czerwcaPoziomy transkryptu mRNA w homogenatach kory od myszy cKO i myszy kontrolnych. W P1 myszy cKO wykazały znacznie wyższe czerwca Poziomy mRNA (*P = 0,01), ale nie w P30 (ns), w porównaniu z grupą kontrolną. Istotność statystyczną określono metodą A Test Manna-Whitneya i B, D, mi nieparzysty T -test. A N = 8 myszy kontrolnych, N = 7 myszy cKO, N = 6 myszy kontrolnych, N = 8 myszy cKO. B N= 8 myszy kontrolnych, N = 8 myszy cKO. D N= 12 myszy kontrolnych, N = 12 myszy cKO, N= 7 myszy kontrolnych, N= 8 myszy cKO. mi N= 8 myszy kontrolnych, N= 7 myszy cKO, N= 11 myszy kontrolnych, N= 13 myszy cKO. Wartości reprezentują średnie ± SEM. nieistotne. Kredyt: Biologia komunikacji(2023). DOI: 10.1038/s42003-023-05612-5
× zamknąć
ROS i markery związane z apoptozą są zmienione w całej korze myszy cKO w porównaniu z grupą kontrolną. Zbadano poziomy mRNA genów związanych z ROS i poziomy białek w homogenatach kory myszy cKO w porównaniu z kontrolą. A Właściwości związane z niedotlenieniem zbadano poprzez pomiar Hif1aPoziomy transkryptu mRNA w homogenatach kory od myszy cKO i myszy kontrolnych. W P1 myszy cKO wykazały znacznie wyższe Hif1aPoziomy mRNA (*P= 0,04), ale nie w P30 (ns), w porównaniu z grupą kontrolną. B Homogenaty całej kory z P1 i P30 cKO w porównaniu z próbkami kontrolnymi poddano immunoblotowi z użyciem przeciwciała Hif1a i nie wykazano istotnej różnicy w poziomach białka. C Reprezentatywne wyniki immunoblotu Hif1a. D Właściwości hamowania ROS zbadano poprzez pomiar Pgc1aPoziomy mRNA w homogenatach kory myszy cKO i myszy kontrolnych. W P1 myszy cKO nie wykazały istotnej różnicy (ns), podczas gdy w P30 myszy cKO wykazywały znacząco niższy poziom ekspresji Pgc1aw porównaniu do kontroli (*P= 0,01). mi Właściwości apoptotyczne badano poprzez pomiar czerwcaPoziomy transkryptu mRNA w homogenatach kory od myszy cKO i myszy kontrolnych. W P1 myszy cKO wykazały znacznie wyższe czerwcaPoziomy mRNA (*P= 0,01), ale nie w P30 (ns), w porównaniu z grupą kontrolną. Istotność statystyczną określono metodą A Test Manna-Whitneya i B, D, mi nieparzysty T-test. A N= 8 myszy kontrolnych, N= 7 myszy cKO, N= 6 myszy kontrolnych, N= 8 myszy cKO. B N= 8 myszy kontrolnych, N= 8 myszy cKO. D N= 12 myszy kontrolnych, N= 12 myszy cKO, N= 7 myszy kontrolnych, N= 8 myszy cKO. mi N= 8 myszy kontrolnych, N= 7 myszy cKO, N= 11 myszy kontrolnych, N= 13 myszy cKO. Wartości reprezentują średnie ± SEM. nieistotne. Kredyt: Biologia komunikacji(2023). DOI: 10.1038/s42003-023-05612-5
Po pierwsze, badacze z Uniwersytetu w Tel Awiwie odkryli, że produkcja i regulacja organelli mitochondrialnych w komórkach nerwowych mózgu (neuronach) zostaje znacznie upośledzona w wyniku delecji genu zwanego Gtf2i, jednego z 25 genów usuniętych w zespole Williamsa.
Wiadomo, że to upośledzenie powoduje niepełnosprawność funkcjonalną komórek nerwowych i może leżeć u podstaw patologii neurorozwojowych, takich jak zespół Williamsa i inne stany związane z genem Gtf2i.
Do odkrycia doszło dzięki wysiłkom zespołu badaczy, na którego czele stoją prof. Boaz Barak z Sagol School of Neuroscience i School of Psychological Sciences na Uniwersytecie w Tel Awiwie oraz Ariel Nir-Sade, dla której te przełomowe badania stanowią jej pracę doktorską.
„W mózgu mamy 100 miliardów komórek nerwowych niezbędnych do utrzymania jego aktywności” – wyjaśnia prof. Barak. „Aby to zrobić, komórki te potrzebują energii. Energia ta jest wytwarzana w organellach mitochondrialnych, dlatego problem z funkcją mitochondriów doprowadzi do problemów z funkcjonowaniem komórki”.
„Dzisiaj rozumiemy, że mitochondria są «winne» za różnorodne patologie neurologiczne, od zaburzeń neurorozwojowych, takich jak zespół Angelmana i autyzm, po choroby neurodegeneracyjne, takie jak choroba Alzheimera i Parkinsona – są to zaburzenia, które obejmują między innymi nieprawidłowe funkcjonowanie mitochondriów.”
W laboratorium prof. Baraka badacze skupiają się na zespole genetycznym zwanym zespołem Williamsa, który wynika z wadliwej ekspresji około 25 genów. Do tej pory nie było jasne, dlaczego komórki nerwowe osób cierpiących na ten zespół ulegają uszkodzeniu – czyli jaka była korelacja między nieprawidłową ekspresją tych genów a wynikającym z tego upośledzeniem funkcji mózgu.
„Zespół Williamsa to stosunkowo rzadki rozwojowy zespół neurogenetyczny” – wyjaśnia Nir-Sade. „Osoby cierpiące na tę chorobę rodzą się z deficytami wielosystemowymi od urodzenia, problemami poznawczymi, motorycznymi i behawioralnymi, ale być może najbardziej wyróżniającą ich cechą są trudności w regulowaniu zachowań społecznych. Dlatego często nazywa się to „syndromem miłości” – osoby te mają tendencję do wykazują znaczne uczucia i silne pragnienie interakcji społecznych.”
Spośród 25 genów, które nie ulegają prawidłowej ekspresji u osób z zespołem Williamsa, badania prof. Baraka i Nir-Sade skupiły się na genie Gtf2i. Gen ten ma kluczowe znaczenie dla zrozumienia tego zespołu, ponieważ koduje czynnik transkrypcyjny – białko odpowiedzialne za regulację wielu innych genów oraz, jak odkryli w swoich badaniach, za regulację ekspresji genów zaangażowanych w mitochondria.
Próbując zrozumieć rolę tego genu w komórkach nerwowych mózgu, badacze z Uniwersytetu w Tel Awiwie zastosowali techniki inżynierii genetycznej w celu porównania struktury mitochondriów w komórkach nerwowych z genem Gtf2i i bez niego.
Zazwyczaj mitochondria współpracują ze sobą w postaci sieci, jednak w przypadku braku genu Gtf2i proces tworzenia sieci nie jest odpowiednio regulowany. W rezultacie tworzenie sieci jest upośledzone, mitochondria mają trudności w funkcjonowaniu, a wewnątrz komórki gromadzą się nieprawidłowe substancje.
„W pierwszym etapie wyodrębniliśmy komórki nerwowe z mózgów zwierzęcych modeli zespołu Williamsa i hodowaliśmy je w hodowli” – mówi Nir-Sade. „Porównaliśmy hodowle normalnych komórek nerwowych z tymi, z których usunięto gen Gtf2i za pomocą inżynierii genetycznej. Zbadaliśmy poszczególne komórki i wykazaliśmy, jak mitochondrium ma trudności z rozwojem i funkcjonowaniem bez tego genu”.
„W drugim etapie, z pomocą laboratorium dr Asafa Marco na Uniwersytecie Hebrajskim w Jerozolimie, chcieliśmy sprawdzić, czy podstawowy mechanizm, który odkryliśmy w kulturach modelowych zwierzęcych, będzie miał zastosowanie również w przypadku ludzi. Zbadaliśmy tkankę mózgową uzyskaną z osób urodzonych z zespołem Williamsa, których mózgi po śmierci zostały przekazane do badań naukowych”.
„Zaobserwowaliśmy, że nasze odkrycia odnoszą się również do ludzkich mózgów: u osób z zespołem Williamsa mitochondria nie rozwijają się i nie funkcjonują prawidłowo, w wyniku czego wewnątrz komórek nerwowych gromadzi się toksyczny materiał, wpływając w ten sposób na ich wydajność”.
„Te odkrycia mają znaczenie kliniczne” – dodaje prof. Barak. „Poprawiają naszą wiedzę na temat tego, co jest potrzebne do poprawy funkcji nerwowych w mózgu – na przykład poprawy funkcji mitochondriów lub zmniejszenia poziomu ekspresji substancji gromadzących się w komórkach nerwowych osób z zespołem Williamsa. Badania biomedyczne poświęcają znaczny wysiłek i zasoby w kierunku zrozumienia chorób mitochondrialnych i w tej dziedzinie dokonuje się znaczny postęp”.
„Jest prawdopodobne, że w przyszłości zostanie opracowany lek poprawiający funkcję mitochondriów w innych schorzeniach, takich jak choroba Alzheimera, a na podstawie naszych badań będziemy wiedzieć, jak dostosować lek również w przypadku zespołu Williamsa, aby poprawić funkcji mitochondriów w tym konkretnym kontekście.”
Praca publikowana jest w czasopiśmie Biologia komunikacji.
Więcej informacji:
Ariel Nir Sade i wsp., Delecja Neuronal Gtf2i zmienia właściwości mitochondrialne i autofagiczne, Biologia komunikacji(2023). DOI: 10.1038/s42003-023-05612-5
Informacje o czasopiśmie:
Biologia komunikacji






