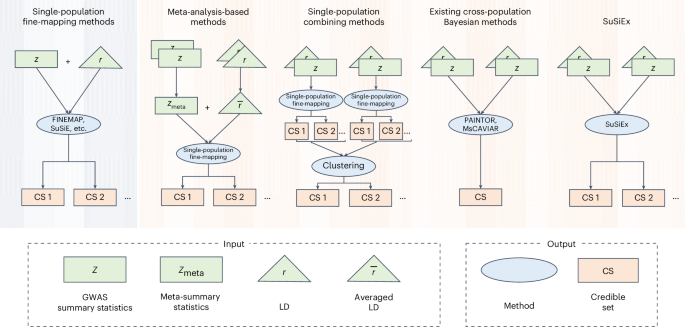
Dokładne mapowanie różnych przodków prowadzi do odkrycia domniemanych wariantów przyczynowych leżących u podstaw złożonych cech i chorób człowieka
Huang, H. i in. Precyzyjne mapowanie loci zapalnej choroby jelit w celu rozwiązania pojedynczego wariantu. Natura 547173–178 (2017).
Maller, JB i in. Bayesowskie udoskonalenie sygnałów asocjacyjnych dla 14 loci w 3 powszechnych chorobach. Nat. Genet. 441294–1301 (2012).
Cortes, A. i in. Identyfikacja wielu wariantów ryzyka zesztywniającego zapalenia stawów kręgosłupa poprzez genotypowanie o dużej gęstości loci związanych z układem odpornościowym. Nat. Genet. 45730–738 (2013).
Trubetskoy, V. i in. Mapowanie loci genomowych wskazuje na udział genów i biologii synaptycznej w schizofrenii. Natura 604502–508 (2022).
Mahajan, A. i in. Wielopłciowe badanie genetyczne cukrzycy typu 2 podkreśla potencjał zróżnicowanych populacji w zakresie odkryć i translacji. Nat. Genet. 54560–572 (2022).
Kanai, M. i in. Dokładne mapowanie metaanalizy jest często błędnie skalibrowane przy rozdzielczości jednowariantowej. Genom komórkowy. 2100210 (2022).
Kanai, M. i in. Wnioski z dokładnego mapowania złożonych cech w różnych populacjach. Preprint at medRxiv (2021).
LaPierre, N. i in. Identyfikacja wariantów przyczynowych poprzez dokładne mapowanie w wielu badaniach. PLoS Genet. 17e1009733 (2021).
Kichaev, G. i Pasaniuc, B. Wykorzystanie danych adnotacji funkcjonalnych w transetnicznych badaniach dokładnego mapowania. Am. J. Hum. Genet. 97260–271 (2015).
Wyss, AB i in. Wieloetniczna metaanaliza identyfikuje loci specyficzne dla przodków i między przodkami dla funkcji płuc. Nat. Komun. 92976 (2018).
Gharahkhani, P. i in. Metaanaliza całego genomu zidentyfikowała 127 loci jaskry otwartego kąta, które mają spójny wpływ na wszystkie grupy etniczne. Nat. Komun. 121258 (2021).
Robertson, CC i in. Analizy precyzyjnego mapowania, transpłciowe i genomiczne pozwalają na identyfikację wariantów przyczynowych, komórek, genów i celów leków w przypadku cukrzycy typu 1. Nat. Genet. 53962–971 (2021).
Wang, G., Sarkar, A., Carbonetto, P. i Stephens, M. Proste, nowe podejście do wyboru zmiennych w regresji, z zastosowaniem do precyzyjnego mapowania genetycznego. JR Stat. Soc. Seria B Stat. Methodol. 821273–1300 (2020).
Sudlow, C. i in. Brytyjski Biobank: ogólnodostępne źródło informacji umożliwiające identyfikację przyczyn szerokiego zakresu złożonych chorób wieku średniego i podeszłego. PLoS Med. 12e1001779 (2015).
Feng, Y.-CA i in. Taiwan Biobank: bogata baza danych badań biomedycznych populacji Tajwanu. Genom komórkowy. 2100197 (2022).
Mitchell, TJ i Beauchamp, JJ Bayesowski wybór zmiennych w regresji liniowej. J. Am. Stat. Assoc. 831023–1032 (1988).
George, EI i McCulloch, RE Podejścia do bayesowskiego wyboru zmiennych. Grzech Stat. 7339–373 (1997).
Google Scholar
Su, Z., Marchini, J. i Donnelly, P. HAPGEN2: symulacja wielu polimorfizmów polimorficznych chorób. Bioinformatyka 272304–2305 (2011).
Auton, A. i in. Globalne odniesienie do zmienności genetycznej człowieka. Natura 52668–74 (2015).
Kichaev, G. i in. Integrowanie danych funkcjonalnych w celu ustalenia priorytetów wariantów przyczynowych w badaniach statystycznego mapowania precyzyjnego. PLoS Genet. 10e1004722 (2014).
Ulirsch, JC i in. Badanie hematopoezy ludzkiej w rozdzielczości pojedynczej komórki i pojedynczego wariantu. Nat. Genet. 51683–693 (2019).
Weissbrod, O. i in. Funkcjonalnie poinformowane mapowanie precyzyjne i poligeniczna lokalizacja dziedziczności złożonych cech. Nat. Genet. 521355–1363 (2020).
Ulirsch, JC Identyfikacja i interpretacja przyczynowych wariantów genetycznych leżących u podstaw fenotypów człowieka. Rozprawa doktorska, Harvard Univ. Graduate School of Arts and Sciences (2022).
Chen, C.-Y. i in. Analiza obejmująca Taiwan Biobank, Biobank Japan i UK Biobank pozwoliła zidentyfikować setki nowych loci dla 36 cech ilościowych. Genom komórkowy. 3100436 (2023).
Li, H. W kierunku lepszego zrozumienia artefaktów w wywołaniach wariantowych z próbek o dużym pokryciu. Bioinformatyka 302843–2851 (2014).
Karczewski, KJ i in. Widmo ograniczeń mutacji określone ilościowo na podstawie zmienności u 141 456 ludzi. Natura 581434–443 (2020).
Benner, C. i in. FINEMAP: efektywny wybór zmiennych przy użyciu danych podsumowujących z badań asocjacyjnych w całym genomie. Bioinformatyka 321493–1501 (2016).
Benner, C. i in. Perspektywy dokładnego mapowania regionów genomicznych powiązanych z cechami przy użyciu statystyk podsumowujących z badań asocjacji w całym genomie. Am. J. Hum. Genet. 101539–551 (2017).
Taliun, D. i in. Sekwencjonowanie 53 831 różnych genomów z programu NHLBI TOPMed. Natura 590290–299 (2021).
Zhou, W. i in. Inicjatywa Global Biobank Meta-analysis: wspieranie odkryć genetycznych w zakresie chorób człowieka. Genom komórkowy. 2100192 (2022).
Pruim, RJ i in. LocusZoom: regionalna wizualizacja wyników skanowania asocjacyjnego w całym genomie. Bioinformatyka 262336–2337 (2010).
Wang, QS i in. Wykorzystanie uczenia nadzorowanego do precyzyjnego mapowania opartego na funkcjonalności cis-eQTLs identyfikuje dodatkowo 20 913 potencjalnych eQTL będących przyczyną choroby. Nat. Komun. 123394 (2021).
Hou, Y. i in. Związana ze schizofrenią specyficzna dla allelu G regulacja w dół FURINA ekspresja miR-338-3p zmniejsza produkcję BDNF. Schizophr. Res. 199176–180 (2018).
Schrode, N. i in. Synergistyczne efekty powszechnych wariantów ryzyka schizofrenii. Nat. Genet. 511475–1485 (2019).
Manichaikul, A. i in. Solidne wnioskowanie o relacjach w badaniach asocjacyjnych całego genomu. Bioinformatyka 262867–2873 (2010).
Chang, CC i in. PLINK drugiej generacji: sprostanie wyzwaniom związanym z większymi i bogatszymi zbiorami danych. Gigascience 47 (2015).
Abraham, G., Qiu, Y. i Inouye, M. FlashPCA2: analiza głównych składowych zestawów danych genotypów na skalę biobanku. Bioinformatyka 332776–2778 (2017).
Han, B. i Eskin, E. Model efektów losowych mający na celu odkrywanie powiązań w metaanalizie badań asocjacyjnych w całym genomie. Am. J. Hum. Genet. 88586–598 (2011).
McLaren, W. i in. Predyktor efektu wariantowego Ensemble. Genom Biol. 17122 (2016).
Willer, CJ, Li, Y. i Abecasis, GR METAL: szybka i wydajna metaanaliza skanów asocjacyjnych w całym genomie. Bioinformatyka 262190–2191 (2010).
Ge, T. i Yuan, K. getian107/SuSiEx: SuSiEx-v1.1.2 (v1.1.2). Zenodo (2024).