Identyfikacja niestabilnych loci powtórzeń CNG w ludzkim genomie: podejście heurystyczne i implikacje dla zaburzeń neurologicznych
Identyfikacja powtórzeń CNG z referencyjnej sekwencji ludzkiego genomu
Poprzez selekcję powtórzeń CNG w całym genomie znaleźliśmy łącznie 15 069 loci (≥ 4 sąsiadujące powtórzenia) (ryc. 1). Powtórzenia CNG były liczne w regionie kodującym i UTR. Ogólnie rzecz biorąc, powtórzenia CAG i CTG były najliczniejsze w różnych regionach genomu (Tabela 1). Opisaliśmy te powtarzające się loci za pomocą ANNOVAR8 i dalej kategoryzował powtórzenia tandemowe na podstawie długości obserwowanej w genomie referencyjnym: Grupa 1, 4–6 powtórzeń; Grupa 2, 7–9 powtórzeń; i grupa 3, > 9 powtórzeń (Tabela 1).
Wybrane powtórzenia tandemowe oceniono in silico pod kątem ich niestabilności i poddano dalszym badaniom przesiewowym w populacji pacjentów z ataksją rdzeniowo-móżdżkową.
Stosując podejście redukcjonistyczne do dalszej analizy, wybraliśmy 52 loci zlokalizowane w regionie CDS lub UTR o długości ciągłych powtórzeń ≥10 (Tabela 2 i Ryc. 2). Powtórzenia zawierające więcej niż 10 jednostek są bardziej podatne na zdarzenia związane z ekspansją9 i powodują spadek aktywności endonukleazy płatkowej-1 (FEN1) na fragmentach Okazaki10. Co więcej, większość patogennych ekspansji powtórzeń trinukleotydów zaobserwowano w regionie kodującym lub UTR, na przykład w SCA1-SCA3 (ekspansja CAG w regionie kodującym), SCA12 (ekspansja CAG w 5′ UTR) i dystrofii miotonicznej (ekspansja CTG w 3’UTR).
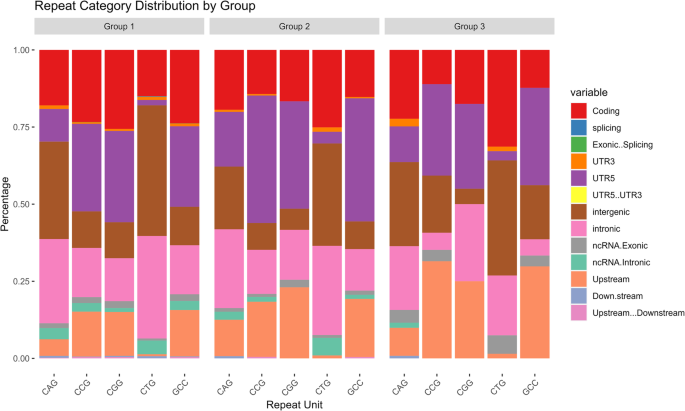
Rozkład powtarzających się kategorii w grupach, pokazujący procent powtórzeń w każdej kategorii, z kolorami reprezentującymi różne zmienne.
Genotypowanie 52 powtórzeń CNG w kontrolnej populacji Indii
Oceniając zmienność długości 52 loci w próbkach kontrolnych, stwierdzono, że 33 loci są stosunkowo stabilne (zmienność długości 1–6 powtórzeń), a 19 loci miało charakter bardziej polimorficzny (zmienność długości 7–23 powtórzeń jednostek). . Te 19 bardziej zmiennych loci powtórzeń (RAI1, UMAD1, GLS, HTR7P1, CNKSR2, MAML3, MED15, MLLT3, USF3, MEF2A, MIR205HG, NCOR2, RPL14, JPH3, MAB21L1, ANKUB1, ERF, GIPC1I EP400) poddano dalszym badaniom przesiewowym w naszej kohorcie pacjentów z ataksją, aby zidentyfikować wszelkie zmiany długości, które mogą być patogenne (ryc. 3).
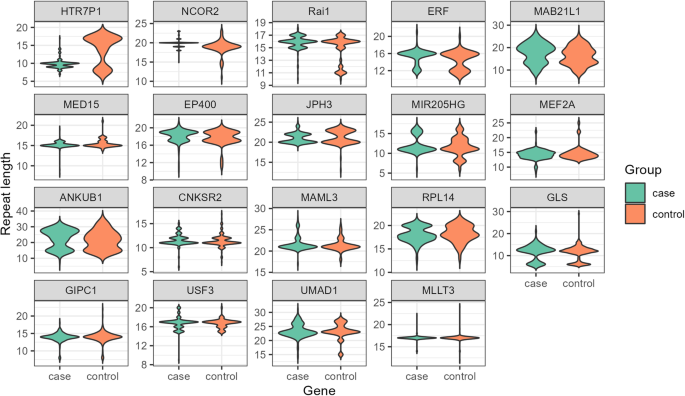
Rozkład powtórzeń docelowych wśród próbek kontrolnych i próbek pacjentów.
The MAB21L1, ANKUB1I GLS geny były wysoce polimorficzne i miały szeroki zakres rozkładów powtórzeń w populacji [modes of repeats (ranges): 13 (8–26), 15 (8–33), and 12 (6–29), respectively]. Geny ANKUB1 I UMAD1 wykazywał dużą liczbę powtórzeń (> 30 powtórzeń) zarówno w grupie przypadku, jak i grupie kontrolnej. Nie zaobserwowano istotnej różnicy w dużym zakresie ekspansji pomiędzy przesiewami przypadku i kontroli (Tabela 2).
Wskaźniki heterozygotyczności (HI, które mierzą liczbę heterozygot w populacji). UMAD1, MAB21L1, ANKUB1, GLSI RPL14 były większe niż 0,7 w obu przypadkach i w grupie kontrolnej. Z drugiej strony, MLLT3 I CNKSR2 były mniej polimorficzne i miały więcej powtórzeń homozygotycznych (HI ≤ 0,1) w obu grupach. Większość docelowych loci mieściła się w zakresie od 0,3 do 0,7, z wyjątkiem ERF, który miał HI mniejszy niż 0,25 we wszystkich próbkach.
Wybór niestabilnych powtórzeń CNG w bazie danych 1000 genomów
Ponieważ związane z chorobą powtórzenia tandemowe są zwykle bardziej polimorficzne w populacji ogólnej, zbadaliśmy polimorficzny charakter tych loci w populacji kontrolnej. W porównaniu z różnymi populacjami kontrolnymi 1000 genomów, tryb powtórzeń i zmienność genu GLS były większe w populacjach afrykańskich i SAS (Tabela 3). MAB21L1 wykazywał większy zakres powtórzeń w populacji EAS. Chociaż niektóre inne loci miały maksymalnie > 20 powtórzeń ekspansji, loci te były jednolite lub mniej zmienne w populacjach. MEF2A był bardzo zmienny, wahając się od 2 do 16 powtórzeń, ale był jednolity w całej populacji. GIPC1 zmienność powtórzeń była mniej powszechna w populacji EUR. Dla MED15 I ERFpowtarzające się dane były dostępne dla bardzo niewielu próbek pacjentów w różnych populacjach. Nie znaleźliśmy żadnych krótkich danych dotyczących powtórzeń tandemowych dla HTR7P1, RPL14, CNKSR2Lub MLLT3 powtórz loci. Nasze powtarzające się dane dla GLS, ANKUB1, EP400, JPH3I RAI1 loci wykazały rozkład bialleliczny, który obserwuje się również w innych głównych populacjach.
Co ciekawe, zaobserwowaliśmy zmienność w zakresach powtórzeń USF3, MEF2A, JPH3, RAI1, ERF, MED15, MAML3I UMAD1 w porównaniu z innymi populacjami świata, ale żadna z różnic nie była istotna według testu rang ze znakiem Wilcoxona (test nieparametryczny). Obie nasze grupy miały stosunkowo mniej powtórzeń dla loci EP400 (Tabela 4). Prawdopodobną przyczyną tej różnicy jest zastosowanie różnych technologii sekwencjonowania; W przypadku danych z projektu 1000 Genomes zastosowano sekwencjonowanie z krótkim odczytem. Chociaż sekwencjonowanie z krótkim odczytem ma swoje zalety, ma również pewną nieodłączną nieefektywność w odniesieniu do wychwytywania powtórzeń dalekiego zasięgu i złożonych regionów genomowych.
Analiza poziomów ekspresji genów zawierających niestabilne powtórzenia
W przypadku wszystkich genów kandydujących porównano ekspresję genów każdego genu w masie tkanek w różnych tkankach za pomocą GTEx11. Analiza wykazała, że CNKSR2, MAB21L1, USF3, RAI1, NCOR2, JPH3, MAML3, EP400I GLS Geny ulegały znacząco silnej ekspresji w mózgu, szczególnie w móżdżku. Wszystkie pozostałe geny, z wyjątkiem MIR205HG, również wykazywał znaczny poziom ekspresji w mózgu (Tabela 2). Ponieważ patogeneza SCA jest związana z mózgiem, wykluczyliśmy to MIR205HG z krótkiej listy genów. W związku z tym zaproponowaliśmy patogeniczność pozostałych 18 genów, które mogą wykazywać fenotyp ataksji.






