MYH6 hamuje postęp nowotworu poprzez zmniejszenie ekspresji KIT w ludzkim raku prostaty
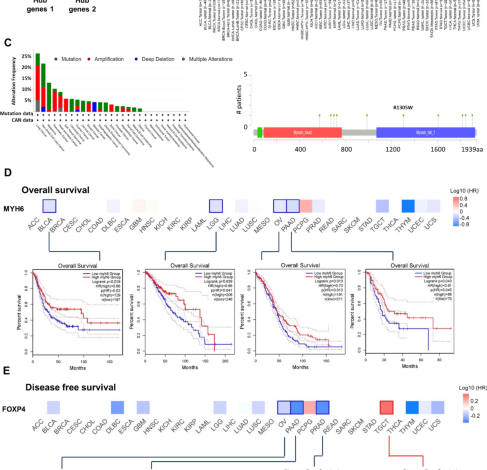
Identyfikacja MYH6 i informacje o danych online
Aby zidentyfikować ważne geny zaangażowane w rozwój raka prostaty, przeanalizowaliśmy geny centralne z naszych poprzednich badań7,8i przeanalizowaliśmy DEGs w bazie danych raka prostaty GDS1439. Poprzez zintegrowaną analizę bioinformatyczną odkryliśmy, że MYH6 był jedynym przecinającym się genem wśród trzech zestawów danych, co potwierdza, że jest naszym genem docelowym. Następnie użyliśmy GEPIA2 do zbadania wzoru ekspresji MYH6 w różnych typach raka i przeanalizowaliśmy dane dotyczące całkowitego przeżycia (OS) i przeżycia bez choroby (DFS), a także wykresy przeżycia związane z MYH6 w różnych typach nowotworów. Profile ekspresji genów raka prostaty i tkanek prawidłowych pobrano odpowiednio z GSE35988 i GDS1439 w bazie danych NCBI-GEO. GSE35988 i GDS1439 to dwie klasyczne publiczne bazy danych dotyczące raka prostaty, zawierające bogactwo danych sekwencjonowania i danych klinicznych od pacjentów z rakiem prostaty. GSE35988 obejmuje dopasowane łagodne tkanki prostaty (n = 28), zlokalizowany rak prostaty (n = 59) i przerzutowy oporny na kastrację rak prostaty (CRPC, n = 35). GDS1439 obejmuje 19 przypadków łagodnych, klinicznie zlokalizowanych guzów raka prostaty, a także przerzutowych i hormonoopornych guzów raka prostaty. W analizie EdgeR został użyty do badania DEGs, jak opisano wcześniej9,10. DEGs zidentyfikowano na podstawie następujących kryteriów: krotność zmiany (FC) ≥ 2 lub ≤ 0,5; wartość p i współczynnik fałszywych odkryć (FDR) < 0,05.
Informacje dla pacjenta
Próbki tkanek od 75 pacjentów z rakiem prostaty, wraz z przyległymi tkankami paranowotworowymi, zostały zebrane do tego badania. Po chirurgicznym usunięciu wszystkie tkanki zostały utrwalone w formalinie. Badanie uzyskało zgodę Komisji Etycznej Uniwersytetu Medycznego w Nankinie, a wszyscy uczestnicy wyrazili pisemną zgodę przed wzięciem udziału.
Immunohistochemia (IHC)
Pozyskaliśmy i poddaliśmy obróbce tkanki sąsiadujące i tkanki raka prostaty zgodnie z wcześniej ustalonymi protokołami11. Wykonano badanie immunohistochemiczne (IHC) zgodnie z opisem w innym miejscu12,13. Krótko mówiąc, preparaty poddano deparafinizacji, a następnie odzyskiwaniu antygenu poprzez ogrzewanie w buforze cytrynianowym o pH 6. Pierwotnymi przeciwciałami były odpowiednio MYH6 (YT6101, Immunoway) i KIT (ab260048, Abcam). Barwienie immunohistochemiczne przeprowadzono przy użyciu techniki streptawidyna-biotyna-peroksydaza, stosując diaminobenzydynę jako chromogen. Pominęliśmy pierwotne przeciwciało lub inkubowaliśmy z niespokrewnionym przeciwciałem, aby uzyskać negatywne kontrole.
Barwienie MYH6 oceniane było niezależnie przez dwóch obserwatorów, z których jeden był patologiem. Na koniec otrzymaliśmy wynik konsensusu dla każdej tkanki. Reakcje MYH6-pozytywne podzielono na cztery poziomy na podstawie intensywności barwienia (0, 1, 2 i 3). Procent komórek MYH6-pozytywnych podzielono na cztery kategorie: 0 (0%), 1 (1–33%), 2 (34–66%) i 3 (67–100%). W przypadku rozbieżności w wynikach rdzenia, wyższy wynik między dwiema tkankami przyjmowano jako wynik końcowy. Ostateczny wynik barwienia uzyskano przez zsumowanie wyników intensywności i procentów. Wzory barwienia klasyfikowano w następujący sposób: 0 dla negatywnego, 1 do 2 dla słabego, 3 do 4 dla umiarkowanego i 5 do 6 dla silnego14.
Hodowla komórkowa
Pozyskaliśmy komórki LNCaP, PC-3 i DU145, dwie klasyczne komórki raka prostaty, z ATCC (Bethesda, USA). Komórki poddano hodowli w podłożu RPMI-1640 uzupełnionym o 10% FBS i antybiotyki (0,1 mg/ml streptomycyny i 100 jednostek/ml penicyliny), zgodnie z protokołem opisanym w poprzedniej literaturze.15Proces uwierzytelniania wszystkich linii komórkowych wykorzystanych w naszym badaniu obejmował profilowanie STR, a także przeprowadzono dokładne testy w celu zapewnienia braku skażenia mykoplazmą.
Budowa stabilnych linii komórkowych
Zajęliśmy się konstrukcjami w sposób opisany wcześniej15,16. W celu skonstruowania pPB-CAG-MYH6-ires-Pac zligowaliśmy pełnej długości MYH6. Stabilne linie komórkowe dla nadmiernej ekspresji MYH6, służące jako kontrola, uzyskano przy użyciu tej samej metodologii, którą opisano wcześniej.15W celu zidentyfikowania i potwierdzenia obecności stabilnych linii komórkowych wykonano analizę Western blot.
Przeciwciała i immunoblotting
Do western blottingu komórki lizowano w 1× buforze ładującym SDS (50 mM Tris–HCl pH 6,8, 10% glicerolu, 2% SDS, 0,05% błękitu bromofenolowego i 1% 2-merkaptoetanolu). Użyto następujących przeciwciał: anty-MYH6 (YT6101, Immunoway), anty-KIT (HY-P80619, MCE), anty-FLAG (20543-1-AP, Proteintech) i anty-ACTIN (20536-1-AP, Proteintech). Immunoblotting przeprowadzono zgodnie z wcześniejszym opisem15. W skrócie, wszystkie białka rozdzielono metodą SDS-PAGE i przeniesiono na membrany poliwinylidenowo-difluorkowe (Millipore). Do wykrywania sygnału użyto przeciwciał wtórnych znakowanych HRP i wzmocnionego systemu chemiluminescencji. Białko uwidoczniono za pomocą systemu obrazowania (Odyssey).
Badanie proliferacji komórek (MTS)
Badanie proliferacji komórek przeprowadzono zgodnie z wcześniej ustalonymi protokołami17Podsumowując, komórki umieszczono w gęstości 4000 komórek na dołek na płytkach 96-dołkowych i hodowano w temperaturze 37 °C przez 6 dni. W określonych odstępach czasu komórki poddawano działaniu 20 μl odczynnika CellTiter 96 AQueous One (Promega) rozcieńczonego w 100 μl podłoża przez 1 h. Następnie przeprowadzono ilościowe oznaczenie komórek przy użyciu czytnika mikropłytek (Biotek).
Analiza i sekwencjonowanie RNA
W naszym badaniu uzyskaliśmy całkowite RNA z komórek przy użyciu odczynnika TRIzol (Invitrogen) i wyeliminowaliśmy wszelkie genomowe DNA przy użyciu DNazy I (TaKara). Następnie RNA zostało skwantyfikowane przy użyciu ND-2000 (NanoDrop Technologies). Do dalszej analizy wybrano tylko próbki RNA o wyjątkowej jakości, spełniające określone kryteria (OD260/280 = 1,8–2,2, OD260/230 ≥ 2,0, RIN ≥ 6,5, 28S:18S ≥ 1,0, > 10 μg), aby skonstruować bibliotekę sekwencjonowania. Biblioteki RNA-seq zostały następnie przetworzone na platformie sekwencjonowania Illumina HiSeq 4000 w Shanghai Majorbio Bio-pharm Technology Co., Ltd (Chiny).
Identyfikacja genów różnicowo ekspresjonowanych (DEG)
Do badania DEGs wykorzystano EdgeR, jak opisano wcześniej9,10. DEGs zidentyfikowano na podstawie następujących kryteriów: krotność zmiany (FC) ≥ 2 lub ≤ 0,5; wartość p i współczynnik fałszywych odkryć (FDR) < 0,05. Te DEGs poddano dodatkowej analizie bioinformatycznej.
Badania migracji komórek
Do przeprowadzenia testów migracji komórek zastosowaliśmy system Transwell (Corning) w 24-dołkowych płytkach do hodowli tkanek, postępując zgodnie z wcześniej ustalonymi protokołami15.
Testy RT-PCR w czasie rzeczywistym
W niniejszym badaniu całkowity RNA został wyizolowany z hodowanych komórek przy użyciu odczynnika TRIzol (Invitrogen). Następnie ten wyekstrahowany RNA został poddany odwrotnej transkrypcji przy użyciu odwrotnej transkryptazy (Fermentas). Ilościowa reakcja PCR w czasie rzeczywistym została przeprowadzona przy użyciu systemu Bio-Rad CFX96, ze względną ekspresją genu znormalizowaną względem GAPDH jako kontroli. Sekwencje primerów wykorzystane w tym badaniu są wymienione poniżej:
-
MYH6-F: GCCCTTTGACATTCGCACTG;
-
MYH6-R: GGTTTCAGCAATGACCTTGCC;
-
GAPDH-F: GGAGCGAGATCCCTCCAAAAT
-
Odwołanie do GAPDH-R:
Badanie wzrostu guza in vivo
Do badania wzrostu guza podskórnie wszczepiliśmy 1 × 106 komórki do sześciotygodniowych samców myszy atymicznych. Każda grupa, obejmująca grupę kontrolną i grupę MYH6-OE, składała się z siedmiu myszy, a wszystkie myszy zostały uśmiercone po 4 tygodniach. Wszystkie guzy monitorowano dwa razy w tygodniu, mierząc rozmiary guzów i rejestrując je jako długość × szerokość2× 0,5 (mm)3). Wszystkie procedury wykorzystania zwierząt zostały zatwierdzone przez Komisję Etyczną Uniwersytetu Medycznego w Nankinie.
Adnotacja funkcjonalna i analiza wzbogacenia ścieżek
Do przeprowadzenia analizy Gene Ontology (GO) i analizy Kyoto Encyclopedia of Genes and Genomes (KEGG) wykorzystaliśmy witrynę internetową Database for Annotation, Visualization, and Integrated Discovery (DAVID)18,19,20,21przy określonym progu istotności statystycznej wartości P < 0,05.
Analiza statystyczna
Wszystkie wyniki przedstawiono jako średnią ± błąd standardowy średnich (SEM). Różnice między grupami oceniano przy użyciu testu Studenta. T-test. Aby przeanalizować związek między ekspresją MYH6 a rokowaniem, wykorzystaliśmy metodę Kaplana-Meiera i test log-rank w GraphPad. Dodatkowo przeprowadziliśmy analizę krzywej charakterystyki operacyjnej odbiornika (ROC) przy użyciu GraphPad. Obszary pod krzywymi (AUC) zostały wykorzystane do oceny przydatności diagnostycznej każdej kombinacji markerów. Wartości AUC przekraczające 0,9 wskazywały na doskonałą skuteczność diagnostyczną, podczas gdy wartości mieszczące się w przedziale od 0,7 do 0,9 sugerowały dobrą skuteczność diagnostyczną. Ponadto współczynnik fałszywych odkryć (FDR) w edgeR i analizie wzbogacenia zestawu genów (GSEA) zostały dostosowane do wielokrotnego testowania za pomocą procedury Benjaminiego-Hochberga w celu kontrolowania FDR22,23Istotność statystyczna została zdefiniowana jako poziom istotności P < 0,05. Wszystkie analizy statystyczne przeprowadzono przy użyciu GraphPad i R 3.3.0.
Zatwierdzenie etyczne
Autorzy twierdzą, że uzyskali odpowiednią zgodę Komisji Etycznej Uniwersytetu Medycznego w Nankinie i postępowali zgodnie z zasadami określonymi w Deklaracji Helsińskiej dla wszystkich badań eksperymentalnych na ludziach lub zwierzętach. Nasze badanie zostało zgłoszone zgodnie z opisem wytycznych ARRIVE (numer referencyjny zatwierdzenia etycznego: 20210329).