Nowy gen, nowe postępy w gangliozydozie
Wyobraź sobie nową fabrykę zbudowaną do produkcji przekąsek. Kilka maszyn współpracuje ze sobą, aby przekształcić ziemniaki w frytki – od mycia, krojenia i solenia ziemniaków po ich gotowanie i pakowanie. Przenośnik taśmowy zabiera przygotowane chipsy do pakowania i wysyłki do magazynów. Jednak maszyny przenośników taśmowych od samego początku są wadliwe, dlatego gdy fabryka rozpoczyna produkcję, przygotowywane są chipsy, ale nic nie jest pakowane ani wysyłane. Wszystkie żetony gromadzą się w jednym miejscu. Z biegiem czasu te stosy wiórów zakłócają działanie maszyn, powodując zamknięcie całej fabryki.
Coś podobnego dzieje się w mózgu Jojo, małej dziewczynki urodzonej z chorobą neurodegeneracyjną, gangliozydozą GM1. Lipidy gromadzą się w jej komórkach mózgowych, ponieważ mechanizm rozkładający te lipidy jest wadliwy.
Gangliozydoza GM1 jest spowodowana mutacjami w genie GLB1 kodującym enzym zwany lizosomalną β-galaktozydazą. Enzym ten rozkłada lipid zwany gangliozydem GM1. Niedobór tego enzymu powoduje gromadzenie się gangliozydu GM1, co skutkuje poważnymi objawami, w tym opóźnieniem wzrostu i rozwoju, powiększeniem wątroby i śledziony, słabym napięciem mięśniowym, nieprawidłowościami szkieletowymi i zaburzeniami wzroku.
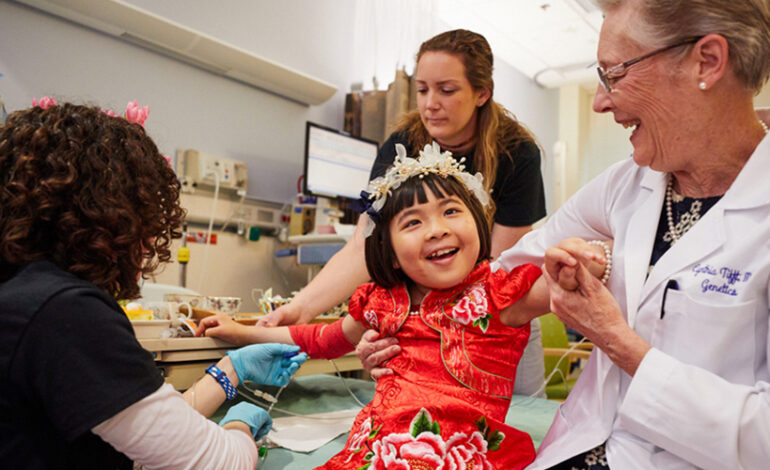
Kiedy Jojo po raz pierwszy przybyła do National Institutes of Health w 2016 roku w wieku 7 lat, potrafiła chodzić, mówić i pisać. W ciągu trzech lat potrzebowała pomocy w staniu, chodzeniu, mówieniu i wykonywaniu niemal wszystkich czynności. W 2019 roku 10-letnia Jojo jako pierwsza osoba została poddana eksperymentalnej terapii genowej z powodu gangliozydozy GM1.
W 2023 roku Laura Allende i jej współpracownicy z NIH pracowali nad chorobą podobną do gangliozydozy GM1: chorobą Taya-Sachsa. Choroba Tay-Sachsa spowodowana jest mutacją w genie HEXA, który koduje enzym odpowiedzialny za degradację gangliozydu GM2. Kiedy Allende zmodyfikowała genetycznie myszy, aby odzwierciedlić mutację u ludzi, odkryła, że myszy miały mniej poważne objawy niż ludzie. To doprowadziło jej zespół do wniosku, że alternatywny gen tworzy szlak obejściowy rozkładający nagromadzone lipidy u myszy, co prowadzi do mniej poważnej postaci choroby.

Ernesto Del Aguila III/Narodowy Instytut Badań nad Genomem Ludzkim
Jojo, pierwsza beneficjentka nowej terapii genowej gangliozydozy GM1, i Cynthia Tifft, autorka badania JLR, świętują przy herbacie.
Naukowcy postulowali, że alternatywny gen, sialidaza NEU3, pomaga w degradacji lipidów. Tak więc, nawet gdy gen HEXA działał nieprawidłowo, szlak obejściowy utworzony przez NEU3 powodował, że myszy miały mniej dotkliwy zespół Tay-Sachsa.
Allende i jej zespół postawili następnie hipotezę, że NEU3 może również łagodzić objawy gangliozydozy GM1. W niedawnym artykule opublikowanym na łamach Journal of Lipid Research, potwierdzili, że NEU3 zapewnia alternatywną drogę degradacji gangliozydów GM1, prowadząc do mniej ostrej postaci gangliozydozy GM1.
W tym celu naukowcy zmodyfikowali genetycznie jedną grupę myszy tak, aby nie posiadała mutacji genu GLB1, a drugą grupę tak, aby nie posiadała genów GLB1 i NEU3. Porównali objawy obu grup i porównali poziom lipidów GM1. Myszy z NEU3 miały mniej poważne objawy, podczas gdy te pozbawione obu genów wykazywały objawy podobne do tych u ludzi.
Dlaczego więc ludzie, którzy również mają gen NEU3, nie mogą wykorzystać szlaku omijającego do degradacji lipidów?
„Przetestowaliśmy ludzkie komórki pod kątem genu NEU3 i powiązanej z nim aktywności enzymatycznej” – powiedział Allende. „Odkryliśmy, że gen ten nie jest tak aktywny u ludzi ze względu na inną sekwencję genetyczną niż ta występująca u myszy”.
Allende uważa jednak, że to odkrycie może nadal pomóc osobom cierpiącym na tę chorobę.
„Wiedza o NEU3 pomoże odkryć nowe potencjalne metody leczenia gangliozydozy GM1 u ludzi” – powiedziała. „Naszym następnym krokiem będzie obserwacja działania NEU3. Jeśli uda nam się włączyć tę alternatywną ścieżkę u ludzi lub sprawić, że enzym NEU3 będzie bardziej aktywny w kierunku degradacji lipidów, będziemy mogli pomóc złagodzić objawy i postęp tej choroby u pacjentów”.
Gangliozydoza GM1 atakuje niemowlęta i może szybko postępować, czasami prowadząc do śmierci w dzieciństwie; im wcześniej choroba zostanie zdiagnozowana, tym większe są szanse na dłuższe i lepsze życie.






