Przypadek ciężkiego zespołu Aicardiego-Goutièresa z homozygotyczną odmianą intronową RNASEH2B
Zespół Aicardiego-Goutièresa (AGS), rzadka postępująca encefalopatia spowodowana nadmiernym zwiększeniem aktywności interferonu (IFN) α, jest pierwszą zidentyfikowaną interferonopatią typu I; interferonopatie typu I to grupa mendlowskich chorób autoimmunologicznych i autozapalnych charakteryzujących się patogennymi polimorfizmami, które zwiększają aktywność sygnalizacji IFN typu I1. RNASEH2B koduje podjednostkę kompleksu RNASEH2, konserwowanego enzymu naprawy DNA, który usuwa nieprawidłowo wstawione monofosforany rybonukleotydów2. patogenny RNASEH2 warianty prowadzą do gromadzenia się metabolitów naprawy DNA, które stymulują szlak cyklicznej syntazy GMP–AMP (cGAS)–stymulatora genów interferonu (STING), co skutkuje nadprodukcją interferonów typu I2Nadmierna odpowiedź autoimmunologiczna powoduje stan zapalny bez infekcji, objawiający się jako objawy wrodzonych infekcji spowodowanych przez Toksoplazmoza gondiiwirus różyczki, cytomegalowirus i wirus opryszczki pospolitej typu 1 i 2 (zespół TORCH) w prenatalnym AGS1Wszyscy zgłaszani pacjenci z AGS są heterozygotyczni pod względem wariantu patogennego c.65-13G > A w RNASEH2Bgen najczęściej mutowany u pacjentów z AGS. Jednak wpływ wariantu c.65-13G > A nie jest znany, a przebieg kliniczny został szczegółowo opisany tylko u jednego pacjenta.
Pacjent był trzecim dzieckiem urodzonym przez rodziców ze Sri Lanki, którzy nie byli spokrewnieni (ryc. 1a). Oboje rodzice i dwoje rodzeństwa nie mieli objawów. Ciężkie zahamowanie wzrostu płodu i małogłowie były zauważalne od drugiego trymestru ciąży. Niemowlę urodziło się przez cesarskie cięcie ze wskazań nagłych z powodu bradykardii płodu w 38. tygodniu i 4. dniu ciąży. Masa urodzeniowa wynosiła 1706 g (−3,89 SD), obwód głowy 27,0 cm (−4,37 SD), a wyniki w skali Apgar wynosiły odpowiednio 7 i 8 punktów po 1 i 5 minucie. Co godne uwagi, upośledzenie oddychania było widoczne od urodzenia i wykonano intubację tchawicy. Leczenie respiratorem kontynuowano przez 5 dni. Wysokoprzepływową kaniulę nosową można było usunąć po 40 dniach, a leczenie tlenem przerwano w 98. dniu życia. Tomografia komputerowa klatki piersiowej wykazała obustronne rozproszone zacienienia przestrzeni powietrznej. Stwierdzono podwyższone poziomy Krebsa von den Lungen-6 (KL-6) (496 U/ml; zakres normy <250 U/ml) i białka surfaktantu-D (SP-D) (149 ng/ml; zakres normy <110 ng/ml), co wskazywało na śródmiąższową chorobę płuc. Niemowlę miało również trudności z karmieniem i biegunkę, którym towarzyszyły zaburzenia równowagi elektrolitowej i kwasica metaboliczna. Udało jej się przybrać na wadze dzięki karmieniu dojajnikowemu mieszanką hydrolizowaną. W ciągu pierwszych kilku miesięcy hipoglikemię hiperinsulinemiczną (poziomy insuliny 4,93–15,50 μU/ml przy niskim stężeniu glukozy we krwi 33–47 mg/dl) kontrolowano za pomocą ciągłego dożylnego podawania glukozy i karmienia dojajnikowego skrobią kukurydzianą, a niedokrwistość z niedoboru żelaza leczono transfuzją czerwonych krwinek i lekami zawierającymi żelazo. Jako profilaktykę przeciwdrobnoustrojową rozpoczęto stosowanie cefakloru (10 mg/kg/dzień) w przypadku obustronnego wodonercza i ciągłej bezgorączkowej pyurii. Zaobserwowano również zaćmę, głuchotę i hepatosplenomegalię, z podwyższonym poziomem transaminaz (aminotransferazy asparaginianowej, 47–212 U/l; aminotransferazy alaninowej, 76–287 U/l). Zmiany przypominające odmrożenia pojawiły się na palcach u stóp w wieku około 2 miesięcy (ryc. 1b). Neuroobrazowanie mózgu wykazało zwapnienie wewnątrzczaszkowe i polimikrogyrię (ryc. 1c, d). Uogólnione drgawki kloniczne obserwowano głównie w ciągu pierwszych kilku tygodni i uległy one poprawie po podaniu fenobarbitalu. Przerost przegrody międzykomorowej (przegroda międzykomorowa przy końcowym rozkurczu, 8,95 mm; z = 3,96) obserwowano od 6 miesiąca życia, chociaż funkcja serca pozostała prawidłowa bez leczenia. U pacjenta rozwinęło się nawracające zapalenie płuc zachłystowe i zmarł w wieku 14 miesięcy z powodu wyraźnie zaostrzonej niewydolności oddechowej i zwiększonej odpowiedzi zapalnej. Dystonia i spastyczność były ciężkie, a wynik AGS, złożony wynik funkcji neurologicznej obliczony na podstawie 11 kluczowych objawów AGS, wynosił 0 przez całe życie pacjenta, bez kontroli głowy, uśmiechu towarzyskiego lub wokalizacji.3.
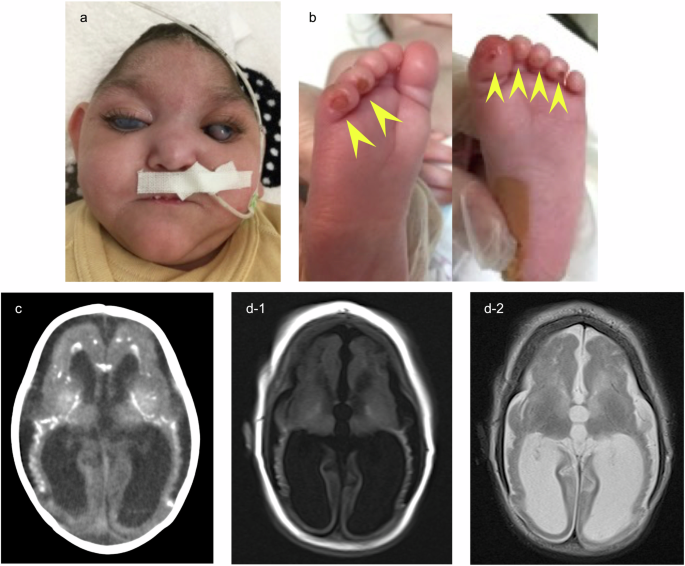
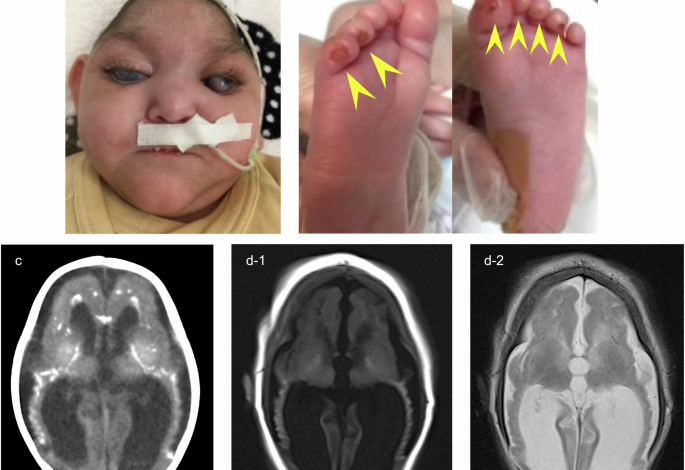
A Pacjent w wieku 12 miesięcy z małogłowiem i obustronną zaćmą. B Zmiany przypominające odmrożenia (żółte groty strzałek) w wieku 2 miesięcy. C Tomografia komputerowa mózgu wykonana w 1 miesiącu życia wykazała zwapnienie jąder podstawy i istoty białej. D MRI mózgu (d-1 T1-ważony, d-2 Badanie T2-ważone wykonane w 4 miesiącu życia wykazało zanik mózgu i polimikrogyrię.
Chociaż podejrzewano zespół TORCH, serologie na toksoplazmozę, różyczkę, cytomegalowirusa i zakażenia wirusem opryszczki były ujemne. Płynu mózgowo-rdzeniowego nie badano; jednak pacjent spełniał pięć głównych kryteriów diagnostycznych niezbędnych do ustalenia klinicznej diagnozy AGS: (i) wczesna encefalopatia z opóźnieniem psychoruchowym, spastycznością, objawami pozapiramidowymi i małogłowiem; (ii) zwapnienia widoczne szczególnie na poziomie jąder podstawy, ale rozciągające się również na okołokomorową istotę białą; (iii) nieprawidłowości istoty białej mózgu; (iv) zanik mózgu; i (v) wykluczenie zakażeń przed-/okołoporodowych, szczególnie zespołu TORCH4W wieku 3 miesięcy przeprowadzono sekwencjonowanie paneli w kierunku dziedzicznych chorób autoimmunologicznych w Kazusa DNA Research Institute i siedem z dziewięciu genów —RNASEH2A, SAMHD1, RNASEH2B, RNASEH2C, TREX1, IFIH1I ADAR—znane z występowania mutacji związanych z AGS zostały przeanalizowane. Chociaż wykryto homozygotyczną odmianę missense w RNASEH2B (NC_000013.11(NM_024570.4):c.895A > G), wariant został sklasyfikowany jako prawdopodobnie łagodny zgodnie z klasyfikacją American College of Medical Genetics and Genomics (ACMG)/Association of Molecular Pathology (AMP)5Badania sekwencjonowania całego eksomu ujawniły kolejny homozygotyczny wariant intronowy w RNASEH2BNC_000013.10(NM_024570.4):c.65-13G > A, str.Glu22Valfs*56który został sklasyfikowany jako patogenny (PVS1 + PS3 + PM2) zgodnie z wytycznymi ACMG/AMP. Sekwencjonowanie całego eksomu oparte na triach rodzinnych potwierdziło, że wariant ten został odziedziczony zarówno od heterozygotycznego ojca, jak i matki (ryc. 2). Nie wykryto żadnych innych patogennych/prawdopodobnie patogennych wariantów. Dlatego u pacjenta zdiagnozowano AGS z powodu obecności patogennego wariantu RNASEH2BBadanie zostało zatwierdzone przez Komisję Etyczną Uniwersytetu Medycznego Jichi (numer zatwierdzenia: A22-022), a rodzice wyrazili pisemną zgodę na udział w badaniu.
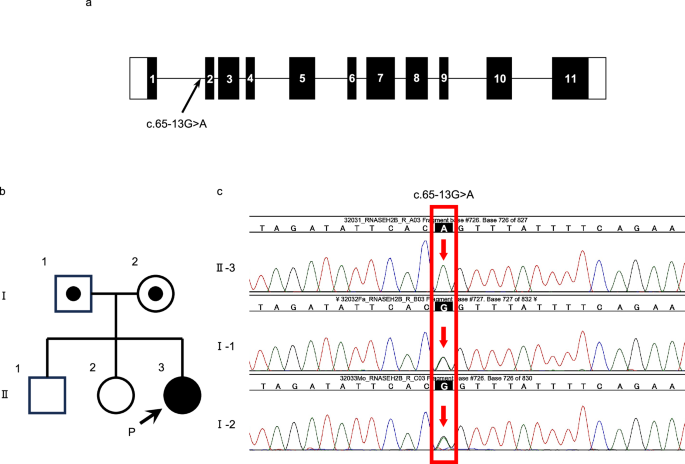
A Schematyczna reprezentacja RNASEH2B gen, z pozycją zidentyfikowanej mutacji. Kolorowe pola z numerami oznaczają eksony, a poziome linie oznaczają introny. B Rodowód rodzinny. C Sekwencjonowanie genomu klinicznego oparte na trio pokazujące wariant c.65-13G > A w RNASEH2B. Elektroforegramy sekwencji DNA homozygotycznego probanta i heterozygotycznych rodziców.
W korelacjach genotypowo-fenotypowych dla AGS początek prenatalny jest najczęściej związany z TREX1 warianty, podczas gdy RNASEH2B warianty są powiązane z pacjentami w wieku niemowlęcym lub z późniejszym początkiem choroby7. RNASEH2B jest genem, który najczęściej ulega mutacji u pacjentów z AGS (36–59%) i wiąże się z niższym wskaźnikiem zachorowalności i śmiertelności niż inne genotypy8,9Patogenny wariant intronowy c.65-13G > A został opisany jedynie jako wariant heterozygotyczny w kilku badaniach6,8,10Częstość występowania tego wariantu w bazie danych agregacji genomu wynosi 0,000003984 (1/251008)11z przewidywaną zmianą białka p.Glu22Valfs*5. Wykazano, że wariant c.65-13G > A powoduje nieprawidłowy splicing, skutkujący zatrzymaniem 11 nukleotydów, co następnie wprowadza przesunięcie ramki odczytu10. Ponadto transkrypt może ulegać rozpadowi mRNA pośredniczonemu przez nonsens ze względu na wprowadzenie kodonu stop. Rice i in.6 zgłosili trzech pacjentów, Crow et al.8 zgłosili trzy rodziny, a Garau i in.10 zgłoszono jednego pacjenta, który zgłosił się z wieloma objawami neurologicznymi w wieku 12 miesięcy (Tabela 1). We wszystkich tych zgłoszonych przypadkach występowały złożone mutacje heterozygotyczne, a wpływ c.65-13G > A nie jest znany. Przebieg kliniczny został szczegółowo opisany tylko w jednym przypadku, który postępował stopniowo w porównaniu z naszym przypadkiem z homozygotyczną zmiennością10,12. Przypuszcza się, że wariant splicingowy w intronie 1, c.65-13G > A, powoduje utratę regionu aminokwasowego na styku RNazy H2B i RNazy H2C, co silnie destabilizuje cały kompleks heterotrimeryczny10Analiza Western blotting wykazała zmniejszenie poziomów wszystkich podjednostek rybonukleazy H2 i zmniejszenie ekspresji rybonukleazy H2B10,13. Chociaż podjednostka RNazy H2A zawiera katalityczny rdzeń RNazy H2, wszystkie trzy podjednostki są wymagane do jej aktywności. Podczas gdy dokładne role podjednostek RNazy H2B i RNazy H2C nie są dobrze poznane, funkcjonalny proliferująco-jądrowy motyw białkowy w RNazie H2B kieruje aktywnością RNazy H2 w replikacji i naprawie14.
Patofizjologia mutacji RNazy H2 prowadzących do fenotypu choroby nie jest w pełni znana. Myszy z niedoborem RNazy H2 wykazują znaczne uszkodzenia DNA, które są zazwyczaj śmiertelne w stadium embrionalnym2,13Wrodzone objawy przypominające zespół TORCH odnotowano tylko u trzech pacjentów z RNASEH2B genotyp9,15: p.A177T/p.Ex9_Ex11del, p.V185G/p.V185G i c.322-3C > G/c.322-3C > G. U pacjenta z homozygotyczną mutacją intronową c.322-3C > G wystąpiły objawy pseudo-TORCH podobne do tych obserwowanych w naszym przypadku15. Młodsze rodzeństwo tej pacjentki również wykazywało głęboką małogłowie i wodniak płodu, a ciąża zakończyła się wewnątrzmaciczną śmiercią płodu w 35. tygodniu ciąży. Reakcja łańcuchowa polimerazy z odwrotną transkrypcją wykazała pominięcie eksonu 5 i wygenerowanie transkryptu poza ramką. Tak więc homozygotyczne mutacje intronowe c.322-3C > G i c.65-13G > A (zidentyfikowane w tym badaniu) są związane z najcięższym fenotypem AGS, co wskazuje na zasadniczą funkcję regionu aminokwasów C-końcowych RNazy H2B. Bezobjawowi rodzice rodzeństwa z homozygotyczną mutacją intronową c.322-3C > G byli spokrewnieni pięć pokoleń temu.15Biorąc pod uwagę rzadkość występowania wariantu c.65-13G > A, istnieje możliwość, że rodzice w naszym przypadku byli również dalekimi krewnymi na Sri Lance.
Podsumowując, homozygotyczna patogenna mutacja intronowa c.65-13G > A w RNASEH2B po raz pierwszy opisano u pacjenta z ciężkim AGS o początku prenatalnym. Przypadek ten ilustruje kluczową rolę regionu aminokwasów C-końcowych RNazy H2B w patogenezie AGS.
Baza danych HGV
Odpowiednie dane z niniejszego Raportu danych są przechowywane w bazie danych zmienności genomu ludzkiego pod adresem