Wykorzystanie ukrytych informacji genetycznych w danych klinicznych dzięki REGLE
Przedstawiamy nową metodę odkryć genetycznych, która może wykorzystać ukryte informacje w wielowymiarowych danych klinicznych. Uczenie się reprezentacji dla odkryć genetycznych w niskowymiarowych osadzaniach (REGLE) jest wydajne obliczeniowo, nie wymaga etykietowania chorób i może zawierać informacje z wiedzy eksperckiej.
Nowoczesne systemy opieki zdrowotnej generują ogromną ilość wielowymiarowych danych klinicznych (HDCD), takich jak pomiary spirogramów, fotopletyzmogramy (PPG), zapisy elektrokardiogramów (EKG), tomografie komputerowe i obrazowanie MRI, których nie można podsumować jako pojedynczej liczby binarnej lub ciągłej (por. „ma astmę” lub „wzrost w centymetrach”). Zrozumienie związku między naszymi genomami a HDCD nie tylko poprawia nasze zrozumienie chorób, ale ma również kluczowe znaczenie dla rozwoju metod leczenia chorób.
HDCH są przechowywane w elektronicznych dokumentach medycznych i dużych projektach biobanków, takich jak UK Biobank w Wielkiej Brytanii, BioBank Japan w Japonii i All of Us w Stanach Zjednoczonych. Projekty te uzyskują zgodę uczestników przed usunięciem danych identyfikacyjnych i udostępnieniem części tego cennego zasobu wykwalifikowanym naukowcom. Celem jest poprawa profilaktyki, diagnostyki i leczenia różnych chorób zagrażających życiu.
Zespół ds. genomiki w Google Research poczynił postępy w wykorzystywaniu HDCD do charakteryzowania chorób lub cech biologicznych, takich jak morfologia tarczy nerwu wzrokowego i przewlekła obturacyjna choroba płuc (POChP). W celu lepszego zrozumienia architektury genetycznej tych konkretnych cech, przeprowadziliśmy wcześniej badania asocjacji w całym genomie (GWAS) dotyczące przewidywań cech generowanych przez nadzorowane modele uczenia maszynowego (ML). Jednak uzyskanie wystarczająco dużych wolumenów danych zawierających etykiety chorób do trenowania nadzorowanych modeli ML nie zawsze jest możliwe. Ponadto proste etykiety chorób nie są w stanie w pełni uchwycić biologii osadzonej w danych bazowych, a nam brakuje metod statystycznych do bezpośredniego wykorzystania HDCD w analizie genetycznej, takiej jak GWAS.
Aby przezwyciężyć te ograniczenia, w artykule „Uczenie się reprezentacji bez nadzoru na podstawie wielowymiarowych danych klinicznych poprawia odkrycia i przewidywania genomiczne” opublikowanym w Genetyka natury, przedstawiamy zasadniczą metodę badania podstawowych czynników genetycznych wpływających na ogólne funkcje organów, które są odzwierciedlone w HDCD. Uczenie się reprezentacji dla odkryć genetycznych w osadzaniach niskowymiarowych (REGLE) jest obliczeniowo wydajną metodą, która nie wymaga etykiet chorób i może uwzględniać informacje z cech zdefiniowanych przez ekspertów (EDF), gdy są dostępne.
Odkrywanie ukrytych informacji w HDCD
Prostym podejściem do badania związku między genami a HDCD jest przeprowadzenie GWAS na każdej współrzędnej danych, np. można badać zmiany w wartości każdego piksela w obrazach medycznych. To podejście jest kosztowne obliczeniowo i ma niską moc wykrywania istotnych powiązań ze względu na wysoką korelację między pobliskimi współrzędnymi i ogromny ciężar wielokrotnego testowania. Bardziej powszechnie stosowanym podejściem jest skupienie się na niewielkiej liczbie cech zdefiniowanych przez ekspertów (EDF) wyodrębnionych z HDCD jako cechach docelowych lub fenotypach GWAS. EDF mogą obejmować klinicznie znane cechy, takie jak wymuszona pojemność życiowa (FVC) lub wymuszona objętość wydechowa w ciągu 1 sekundy (FEV1) w przypadku spirogramów, jak zilustrowano w poprzedniej pracy. Chociaż te EDF są ważnymi cechami odkrytymi przez ekspertów przedmiotowych, postawiliśmy hipotezę, że mogą one nie wychwytywać kompleksowo sygnałów zakodowanych w HDCD, a zatem uruchomienie GWAS na nich może nie wykorzystać pełnego potencjału HDCD.
REGLE ma na celu przezwyciężenie tych ograniczeń przy użyciu modelu autokodera wariacyjnego (VAE). Metoda składa się z trzech głównych kroków: (1) nauczenie się nieliniowej, niskowymiarowej, rozplątanej reprezentacji (tj. kodowania lub osadzenia) HDCD za pomocą VAE; (2) przeprowadzenie GWAS na każdej współrzędnej kodowania; i (3) użycie poligenicznych wyników ryzyka (PRS) ze współrzędnych kodowania jako wyników genetycznych ogólnych funkcji biologicznych, a następnie potencjalne połączenie tych wyników w celu utworzenia PRS dla określonej choroby lub cechy (przy założeniu niewielkiej liczby etykiet chorób). Co godne uwagi, REGLE umożliwia również opcjonalne uwzględnienie odpowiednich EDF w danych wejściowych do dekodera w zmodyfikowanej architekturze VAE, tak aby koder był zachęcany do nauki tylko sygnałów resztkowych niereprezentowanych przez EDF.
Wykrywanie nowych loci genetycznych dla funkcji płuc i układu krążenia
Demonstrujemy możliwości REGLE przy użyciu dwóch wielowymiarowych klinicznych modalności danych: spirogramów mierzących funkcję płuc i PPG mierzących funkcję układu sercowo-naczyniowego. Oba mogą być zbierane w sposób nieinwazyjny, stosunkowo niedrogi w klinikach lub z urządzeń noszonych przez konsumentów, a istnieją dobrze znane cechy dla obu modalności (np. FEV1 lub FVC dla spirogramów i obecność lub lokalizacja wcięcia dykrotycznego dla PPG). W porównaniu do badań asocjacyjnych całego genomu dotyczących cech spirogramu i PPG o tym samym wymiarze, badania REGLE nad poznanymi kodowaniami odzyskują większość znanych loci genetycznych powiązanych z funkcją płuc i układu krążenia, a także wykrywają dodatkowe loci (np. 45% bardziej znaczących loci dla PPG). Jeśli loci te zostaną zweryfikowane w dalszej analizie i eksperymentach laboratoryjnych, mają potencjał, aby stać się nowymi celami leków.
Poprawa wyników ryzyka genetycznego
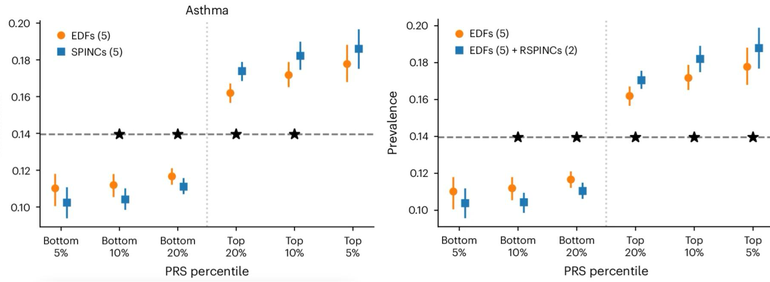
Wynik ryzyka poligenicznego (PRS) to podsumowanie szacowanych efektów wielu wariantów genetycznych na konkretną cechę, reprezentowane jako pojedyncza liczba. PRS utworzone przez badania asocjacji w całym genomie na osadzonych REGLE można łączyć, używając tylko niewielkiej liczby etykiet chorób, aby wygenerować PRS dla tej konkretnej choroby (krok 3 powyżej). Zaobserwowaliśmy, że PRS funkcji płuc utworzone z kodowania spirogramów poprawiły przewidywania POChP i astmy w porównaniu z istniejącymi metodami (takimi jak PRS generowane przez cechy zdefiniowane przez ekspertów, PCA i dopasowanie spline) i stratyfikowały grupy ryzyka skuteczniej niż PRS cech na obu końcach spektrum ryzyka. Zaobserwowaliśmy statystycznie istotną poprawę w wielu metrykach (AUC-ROC, AUC-PR i korelacja Pearsona) w wielu niezależnych zestawach danych (COPDGene, eMERGE III, Indiana Biobank i EPIC-Norfolk) dla astmy i POChP, jak pokazano poniżej.
Porównanie kodowania spirogramów (SPINC) i kodowania spirogramów resztkowych (RSPINC) PRS z PRS o cechach zdefiniowanych przez ekspertów w astmie rozpowszechnienie. Pozioma linia przerywana pokazuje całkowitą częstość występowania. Niższy jest lepszy dla dolnych percentyli; wyższy jest lepszy dla górnych percentyli. * oznacza statystycznie istotną różnicę.
Podobnie, PRS pochodzące z osadzeń REGLE PPG poprawiają prognozy nadciśnienia i skurczowego ciśnienia krwi (SBP). Współpracowaliśmy z współpracownikami akademickimi, aby ocenić nadciśnienie i SBP PRS generowane przez kodowania PPG i cechy PPG w trzech niezależnych zestawach danych (COPDGene, eMERGE III i EPIC-Norfolk) oprócz zestawu testowego z UK Biobank. Zaobserwowaliśmy stały trend poprawy w wyniku korzystania z PRS z kodowań PPG w porównaniu z tymi z cech zdefiniowanych przez ekspertów, zarówno dla nadciśnienia, jak i SBP w wielu zestawach danych.
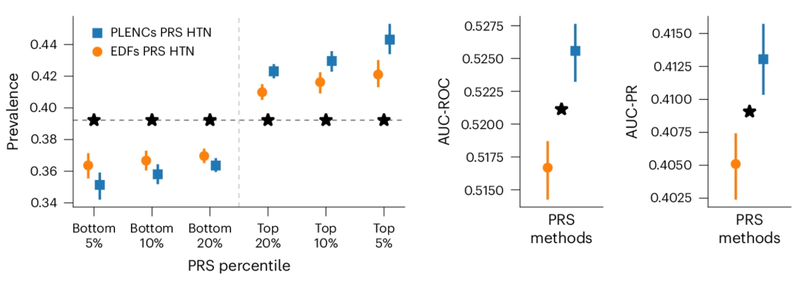
Porównanie kodowania PPG (PLENC) PRS dla nadciśnienia (HTN). Występowanie, AUC-ROCI AUC-PR są obliczane. * oznacza różnicę statystycznie istotną.
Częściowo interpretowalne osadzenia
Wykorzystanie generatywny natura REGLE, badaliśmy wpływ współrzędnych kodowania na kształt spirogramu, ustalając wartości cech zdefiniowanych przez ekspertów i zmieniając jedną współrzędną kodowania, utrzymując pozostałe na poziomie zerowym. Następnie generujemy odpowiadające im spirogramy, używając tylko części dekodującej wytrenowanego modelu. Typowy spirogram przepływu i objętości składa się z dwóch odrębnych części: (1) stosunkowo krótkiej części, aby osiągnąć szczytowy przepływ, gdzie przepływ wzrasta monotonicznie wraz ze wzrostem objętości, oraz (2) głównej części spirogramu, gdzie przepływ maleje monotonicznie. Poniższy rysunek pokazuje, że zmiana pierwszej współrzędnej oznacza poszerzenie lub zwężenie drugiej części (ujemne nachylenie), przy jednoczesnym zachowaniu pierwszej części stosunkowo stałej. W rzeczywistości wklęsłość drugiej części krzywej jest dobrze znana pulmonologom jako listwa przypodłogowawskaźnik niedrożności dróg oddechowych, który nie jest dobrze reprezentowany przez standardowe wskaźniki EDF.
Wniosek
Zaprezentowaliśmy REGLE, metodę uczenia bez nadzoru, która wykonuje analizę genetyczną, ulepszone odkrywanie nowych loci i przewidywanie ryzyka, aby przezwyciężyć ograniczenia z naszej poprzedniej pracy nad fenotypowaniem opartym na uczeniu maszynowym. Uczenie bez nadzoru reprezentacji HDCD do odkrywania genomów jest atrakcyjne ze względu na trudność ręcznego odkrywania EDF na dużą skalę. Struktura REGLE wspiera również zasadnicze wykorzystanie takich cech w modelowaniu poprzez modyfikację tradycyjnej architektury VAE. Demonstrujemy REGLE w dwóch modalnościach danych klinicznych, spirogramach i PPG, które można rutynowo mierzyć w warunkach klinicznych, a także można je mierzyć pasywnie i nieinwazyjnie za pomocą smartfonów lub urządzeń noszonych.
REGLE zapewnia mechanizm identyfikacji genetycznych wpływów na funkcjonowanie organów w przypadku braku oznaczonych danych i naturalnie przyznaje się do włączania cech eksperckich do modelu. Zapewnia również metodę tworzenia PRS specyficznych dla choroby i cechy z bardzo małą liczbą etykiet. W miarę jak biobanki z bogatym obrazowaniem, monitorowaniem aktywności, dokumentacją medyczną i sparowanymi danymi genetycznymi nadal się rozwijają, przewidujemy, że ta lub podobne metody będą coraz częściej stosowane w celu dalszego wyjaśnienia genetycznych podstaw ludzkich cech i chorób.
Podziękowanie
Ta praca jest wspólnym dziełem wielu współpracowników i instytucji. Dziękujemy wszystkim współpracownikom: Justinowi Cosentino, Babakowi Behsazowi, Yuchenowi Zhou, Zachary’emu R. McCawowi, Howardowi Yangowi, Andrew Carrollowi, Cory’emu Y. McLeanowi (Google), Davinowi Hillowi (Northeastern University), Tae-Hwi Schwantes-An, Dongbingowi Lai (Indiana University), Johnowi Batesowi (Verily), Brianowi D. Hobbsowi, Michaelowi H. Cho (Brigham and Women’s Hospital & Harvard Medical School), Robertowi Lubenowi, Anthony’emu P. Khawaji (Moorfields Eye Hospital & University College London). Dziękujemy również Nickowi Furlotte za zrecenzowanie manuskryptu, Gregowi Corrado i Shravya Shetty za wsparcie oraz Annisah Um’rani za pomoc w logistyce publikacji.